El Reglamento europeo cumple veinte años: de los 8 medicamentos existentes para enfermedades poco frecuentes en el año 2000 se ha pasado a los 169 disponibles hoy
Uno de los desafíos en este campo es garantizar el acceso de los pacientes a los tratamientos; laboratorios ya participan en acuerdos basados en resultados en salud
![]()
octubre 2020
A principios de 2000, el Diario Oficial de la Unión Europea publicaba el Reglamento sobre medicamentos huérfanos para abordar las necesidades de los 30 millones de pacientes europeos que viven con una de las denominadas enfermedades raras o poco frecuentes. Antes de su aprobación y puesta en marcha, la investigación terapéutica en esta área era limitada, fundamentalmente por el pequeño número de pacientes al que afecta cada patología y la complejidad y el riesgo asociados al desarrollo de nuevos medicamentos.
Era necesario incentivar y alentar la investigación, y el reglamento sobre fármacos huérfanos estimuló el desarrollo de nuevos tratamientos para enfermedades que no tenían esperanza. Las cifras avalan esta afirmación: antes del año 2000 la Agencia Europea de Medicamentos (EMA) tenía autorizados solamente ocho productos para enfermedades raras, y en 2019 ya eran 169 los medicamentos disponibles para casi un centenar de patologías diferentes, junto a más de 2.100 designaciones huérfanas (es decir, nuevas indicaciones para patologías poco frecuentes autorizadas por la EMA a medicamentos ya existentes).
Del mismo modo, de los casi 30 medicamentos con nuevos principios activos aprobados el año pasado por la EMA, siete fueron para terapias consideradas huérfanas, lo que supone que uno de cada cuatro nuevos fármacos están destinados a pacientes con enfermedades poco frecuentes. En este sentido, 2018 se convirtió en el año más prolífico, con 22 nuevos tratamientos, un hito en este terreno.
Asimismo -y quizá el dato más importante por el horizonte que representa-, en sólo una década los proyectos de investigación clínica en este ámbito han crecido un 88%, según los datos que maneja la Federación Europea de la Industria Farmacéutica (Efpia).

Redes de referencia, planes nacionales, más empresas…
Europa se ha beneficiado enormemente del Reglamento sobre medicamentos huérfanos, porque, además de todo lo anterior, ha logrado una mayor comprensión de las enfermedades raras -esencial para el desarrollo de nuevos tratamientos-; se han creado 24 redes de referencia; 23 Estados miembro han puesto en marcha planes nacionales de este tipo de enfermedades; se han creado 220 empresas, responsables del 51% de los medicamentos huérfanos que se han aprobado en Europa, y se ha impulsado la labor de los pacientes y el movimiento asociativo, que permite, a su vez, mejorar la comunicación y el conocimiento sobre estas patologías de los profesionales sanitarios, investigadores, compañías farmacéuticas y autoridades sanitarias.
La norma prevé, entre otras medidas de estímulo, mayor agilidad en el proceso de aprobación, un protocolo de asistencia y asesoramiento específico por parte de la EMA y ayudas financieras para la realización de ensayos clínicos, además de un periodo de exclusividad en el mercado tras la aprobación del nuevo medicamento (10 años, o bien 12 si el fármaco se autoriza para uso pediátrico).
“Seguir garantizando un marco regulatorio estable y predecible, con incentivos para la industria farmacéutica, apoyará aún más la investigación y desarrollo de nuevos tratamientos tan necesaria para las enfermedades poco frecuentes”, valora lciar Sanz de Madrid, directora del Departamento Internacional de Farmaindustria.
“El Reglamento europeo de medicamentos huérfanos ha brindado esperanza a las personas que viven con estas enfermedades, y los incentivos que aporta a los laboratorios han transformado la vida de los pacientes y de sus familias, mejorando los resultados en salud y contribuyendo a la economía de toda la Unión Europea. Queda mucho por avanzar y el camino es largo, pero esta es la dirección correcta”, añade.
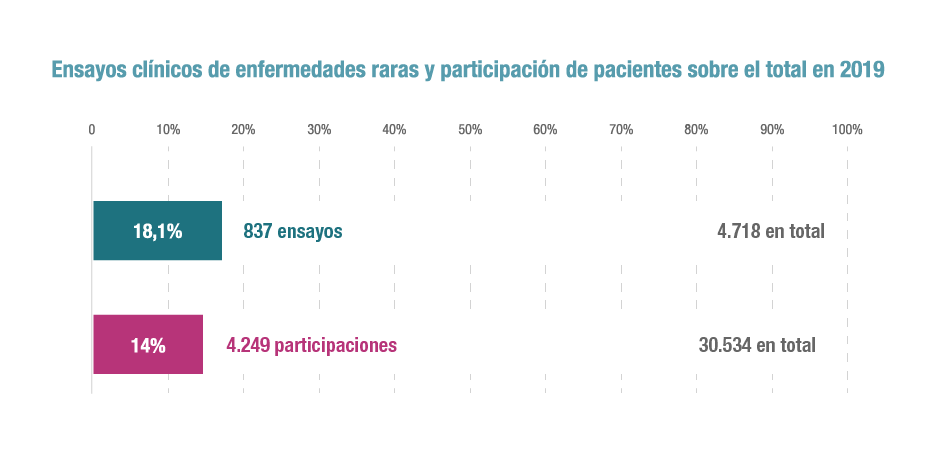
Y es que la norma ha tenido un claro reflejo en el escenario español de investigación clínica. Según los datos del Registro Español de Estudios Clínicos (REEC), que coordina la Agencia Española de Medicamentos y Productos Sanitarios (Aemps), de los más de 4.000 ensayos en curso el año pasado 837 están focalizados en patologías poco frecuentes, con una participación de más de 4.200 pacientes.
Entre 5.000 y 8.000 patologías distintas
Se trata de patologías, en su mayoría, de origen genético, que emergen en la infancia y suponen una grave carga de enfermedad para las personas que las padecen. Son enfermedades complejas, científicamente desafiantes y heterogéneas en la naturaleza. Se estima que existen en todo el mundo entre 5.000 y 8.000 distintas, raras o muy raras.
Hoy, el 95% de ellas aún necesitan opciones de tratamiento. Esto se debe principalmente a la diversidad que existe de enfermedades, a que el colectivo de pacientes afectados por cada una de ellas es pequeño y disperso, a la falta de biomarcadores y a la dificultad para el diagnóstico y abordaje clínico, así como a la citada complejidad en el descubrimiento de nuevos tratamientos. Sin embargo, “cuando se inicia la investigación clínica en un medicamento huérfano todos los pacientes de esa patología se benefician porque se comporta como una palanca de cambio en la asistencia de la enfermedad”, afirma Sanz de Madrid.
El reglamento europeo también ha supuesto una revolución en el diseño de los ensayos clínicos, que son cada vez más adaptativos, racionales y optimizan el proceso. Los ensayos en estas enfermedades suelen ser necesariamente de dimensiones reducidas, lo que hace su planificación más compleja. Por tanto, estos diseños son una alternativa interesante para abreviar el proceso de desarrollo sin afectar a la validez o la eficacia. Además, los tratamientos ineficaces se pueden identificar en etapas más tempranas y permiten un uso más eficaz de los recursos.
Los biomarcadores también permiten reducir los tiempos y costes de los nuevos ensayos clínicos. En este sentido, la introducción de la genómica en la investigación ha provocado un cambio de paradigma en el modelo de investigación de la industria farmacéutica. Todo ello ha implicado nuevos métodos y fórmulas para mejorar la eficiencia de la I+D de los medicamentos huérfanos, es decir, que mejoren el éxito con menor coste y tiempo de desarrollo.

aún no tienen una respuesta terapéutica
Innovación sostenible y acceso de los pacientes al tratamiento
Desde Efpia se ha hecho un llamamiento a las autoridades sanitarias a formular políticas que apoyen incentivos sólidos para la investigación y el desarrollo de medicamentos huérfanos que den esperanza a los pacientes que viven con enfermedades poco frecuentes. “No es momento de poner en peligro un reglamento probado y efectivo, sino de incentivar aún más la investigación de nuevos tratamientos para pacientes que actualmente no tienen ninguna otra opción terapéutica”, añaden desde la Federación ante el anuncio de la Comisión de Europea de revisar la normativa en el marco de la futura Estrategia Industria Farmacéutica de la UE.
“Además de nuestra responsabilidad colectiva de encontrar nuevas opciones de tratamiento para pacientes que sufren alguna de las miles de patologías raras que existen, el Reglamento de medicamentos huérfanos está llamado a desempeñar un papel en la futura Estrategia Industrial de la UE, ayudando a promover la innovación sostenible”, subrayan desde Efpia.
El otro gran desafío reside en asegurar el acceso de los pacientes a los tratamientos innovadores garantizando la sostenibilidad de los sistemas sanitarios nacionales. También aquí ha habido progresos, y las compañías farmacéuticas ya participan en acuerdos innovadores con las autoridades sanitarias de los Estados miembro que están permitiendo un acceso ágil y que se vinculan a los resultados en salud, de modo que, por ejemplo, sea la compañía farmacéutica y no la Administración la que corra con el coste del medicamento en aquellos pacientes en los que no se logre el resultado esperado.
Es evidente que hoy existe una mayor sensibilización de las compañías farmacéuticas hacia acciones concretas para este tipo de patologías -incluso han creado nuevos departamentos ad hoc– y una mayor concienciación social, pero aún queda mucho por avanzar. La creación de registros de pacientes, la importancia de compartir datos y una mayor colaboración público-privada ayudarían también en este viaje que inició hace veinte años el Reglamento europeo de medicamentos huérfanos, que requiere de la cooperación global de todos los agentes para seguir investigando y, por tanto, ofreciendo esperanza para los pacientes en las próximas décadas.

